Novel Compound Designed to Increase Protein Expression in SCN1A loss of function Mutations
•More than 700 mutations of the Scn1a gene have been identified making this the most commonly mutated gene in human epilepsy.
•More than 50% of these mutations result in a truncated protein clearly demonstrating haploinsufficiency of SCN1A as a cause of Dravet Syndrome. This means the mutation leads to a loss of function due to a premature stop codon (frameshift, nonsense, splice site, deletion, microdeletion)
•Analysis of 333 patients showed a mutation spectrum, including whole gene deletions, which argues in favor of loss of function as the main mechanism responsible for Dravet syndrome.
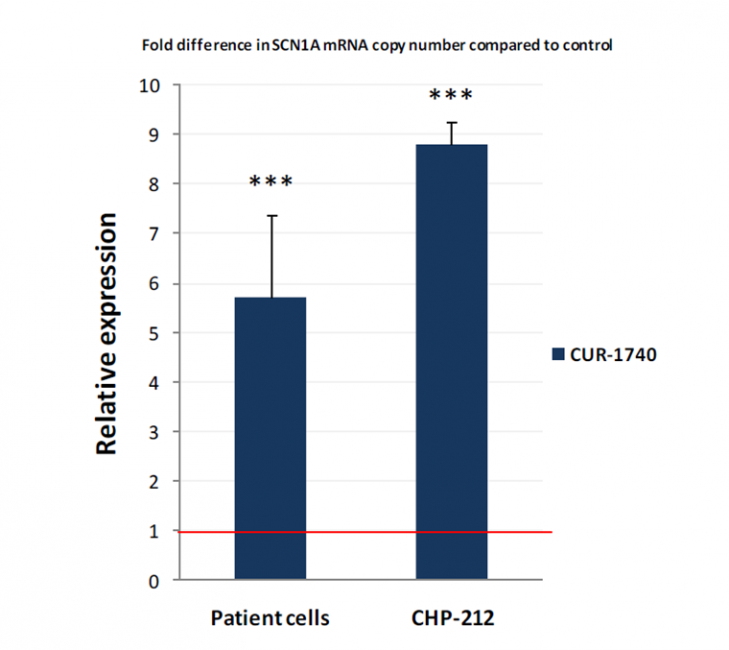
CUR-1740 increased SCN1a expression in an SCN1a mutated human fibroblast by > 8x